Nucleation marks the onset of phase transitions – for instance, when water vapor begins to condense into liquid droplets. These processes are critical across many scientific domains, especially in atmospheric science.
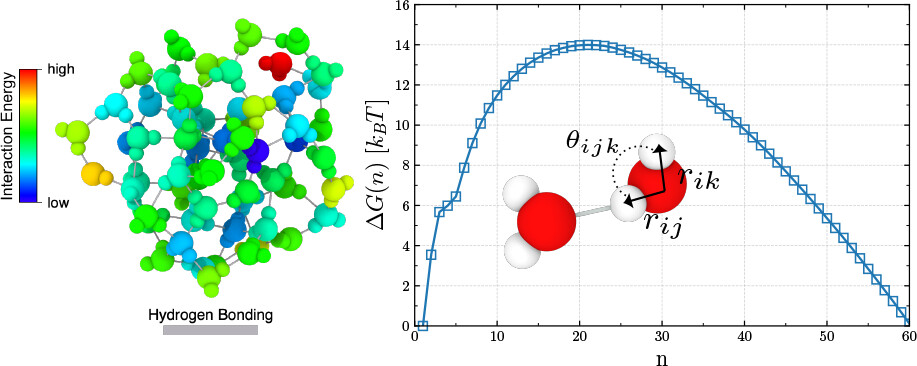
In a previous project, we used molecular dynamics to parameterize an interatomic potential that accurately reproduces liquid water properties such as density, diffusion, pH, and local force features. However, applying the same approach to nucleation is far more challenging: nucleation events are rare, and capturing them directly with molecular dynamics would require infeasible computational effort.
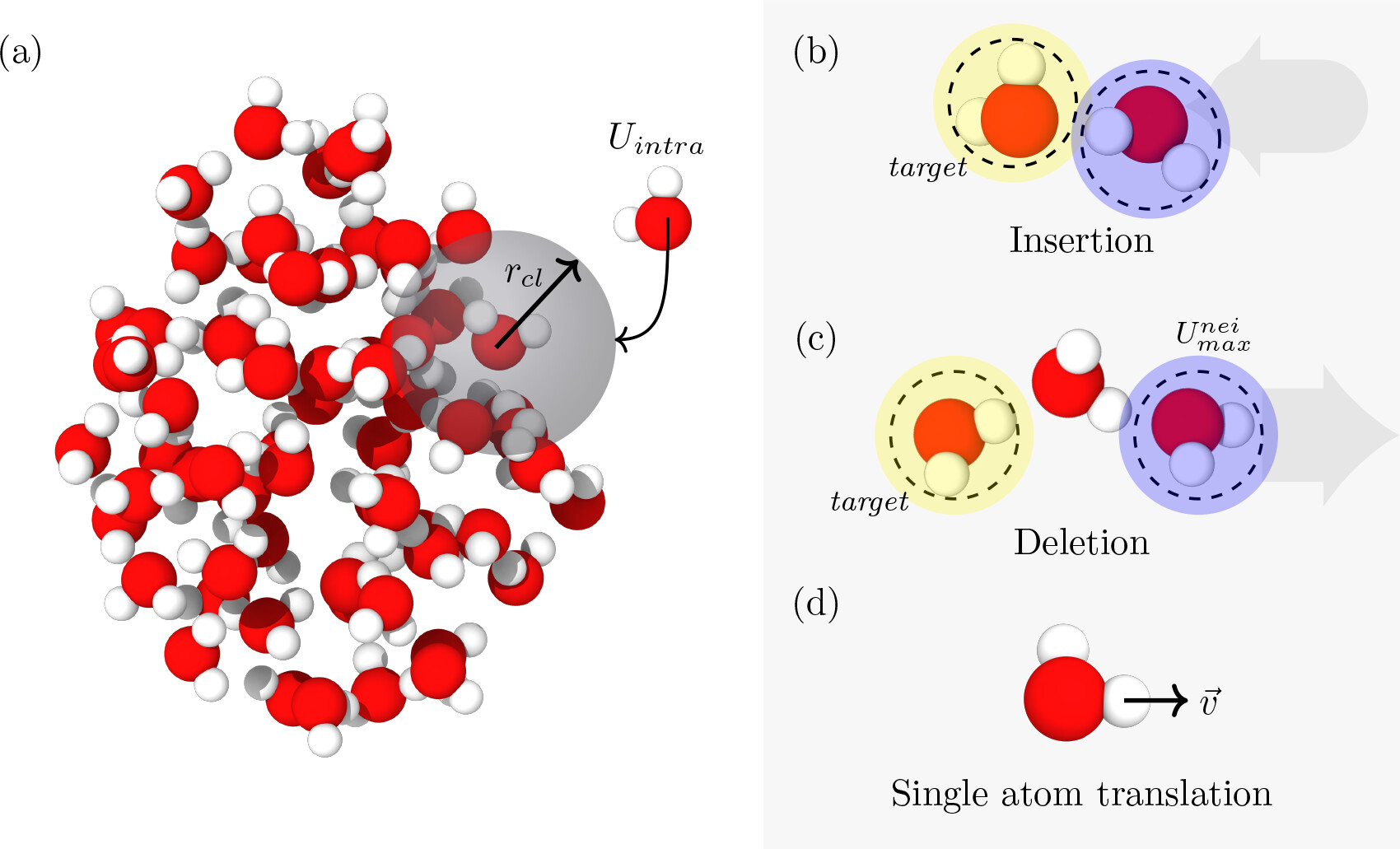
We introduce two new Monte Carlo moves for inserting and deleting reactive molecules in/from droplets.
In our new paper, we tackle this problem using biased Monte Carlo sampling. Building on the aggregation-biased Monte Carlo (AVBMC) method, we extend it to handle reactive water systems. This allows us to estimate key thermodynamic quantities like the saturation vapor density and surface tension for a given potential—both crucial for simulating accurate phase behavior.
Read the full article here:
Camposano, Nordhagen, Malthe-Sørenssen, and Sveinsson — Journal of Chemical Theory and Computation, Article ASAP, DOI: 10.1021/acs.jctc.5c00722
Feel free to drop a comment or question below if you have thoughts or experiences you’d like to share.
